 Dr. Norbert Pöllinger Glatt Pharmaceutical Services GmbH & Co.KG
Dr. Norbert Pöllinger Glatt Pharmaceutical Services GmbH & Co.KG
A drug’s efficacy is linked inextricably to its solubility. Without it, drugs are less effective or not effective at all, because they don’t achieve therapeutic concentration in the patient’s body. That’s an issue, because around 70% of newly discovered drug candidates and approximately 40% of marketed oral drugs are considered practically insoluble.1 Whenever there is a significant difference between the solubility of the active pharmaceutical ingredient (API) and the quantity to be absorbed, measures need to be taken. Two available technologies offer some promise: amorphous solid dispersions (ASD) and solid dispersions (SD) including solubilizers.
The multiparticulate formulation approach—including both ASD and SD—can on one hand ensure adequate solubility and bioavailability. On the other, it can improve patient acceptance in terms of the medicine’s taste and size. The active ingredient often is pressed into large tablets. But that approach can be problematic for both children and older adults, who may have issues with taste or difficulty swallowing large tablets. Multiparticulate strategies—such as pellets and micropellets—provide a more palatable alternative.
The following three approaches apply that strategy:
1. Multiparticulate Amorphous Solid Dispersions
A common approach to improve the solubility is to formulate the drug as amorphous solid dispersions (ASD) with suitable polymer candidates.2 ASDs have been used for many years as a basic technology to improve solubility and bioavailability of poorly soluble dihydropyridine compounds such as Nifedipine and Nimodipine, using PVP 25 as the functional polymer.5

In an amorphous solid dispersion, the basically crystalline active ingredient is dispersed within a polymer matrix in an amorphous form (Figure 1). With the drug in an amorphous form, no energy is required to break a crystal lattice. For this reason, relative to the crystalline form, the amorphous form of many poorly water-soluble drugs can achieve substantially higher apparent water solubility and markedly faster dissolution. ASDs are also known to result in higher membrane flux due to a higher supersaturation and thus, improve bioavailability. The hydrophilic polymer in an ASD also improves the wettability of lipophilic drugs. In addition, ASDs can include surfactants in order to further enhance drug release and stability.6
Hot-melt extrusion may not be a feasible technology to transfer thermo-sensitive compounds into ASDs; heating may chemically decompose or deactivate the API.3,4
To alternatively produce ASDs with a solvent evaporation method, the active compound is dissolved in an organic solvent, e.g. ethanol, together with a polymer, e.g. polyvinylpyrrolidone (PVP) (Figure 2). Polymers frequently used to form ASDs are polyvinylpyrrolidone (PVP K25 and K30), polyvinyl-pyrrolidone/ vinyl acetate copolymer (Kollidon VA 64), polymethacrylate derivates (e.g. Eudragit types), hydroxypropyl methylcellulose (HPMC) and hydroxypropyl methylcellulose acetate succinate (HPMC AS).
When mixtures of solvents are used, azeotrope systems are preferred as they allow for rapid evaporation and effective production.6 As soon as the organic solvent is evaporated, an amorphous solid dispersion of API and polymer is achieved – provided that a feasible polymer quality and API/polymer ratio has been applied. Evaporation of the organic solvent(s) leads to a coprecipitation forming an ASD layer on starter beads or ASD matrix pellets.
Now the question is: which is the best method for evaporating the solvent? What are the advantages and disadvantages?
The answer depends, in part, on the delivery method. For the preparation of pellets and micropellets including ASD, a fluidized bed layering technology and a matrix pelletization technology can be advantageously applied (Figure 2).
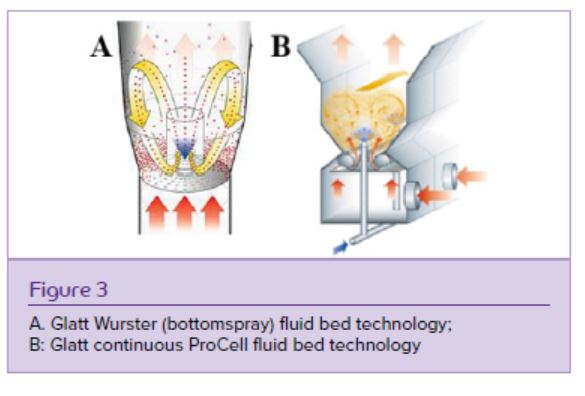
To manufacture ASD layered pellets, the drug-polymer solution is sprayed on inert cores using the Wurster (bottomspray) fluid bed technology (Figure 3A). To provide ASD matrix pellets, alternatively, it can be directly processed to matrix pellets or micropellets using the continuous Glatt fluidized bed systems MicroPx or ProCell (Figure 3B). With these technologies, ASD pellets and micropellets are generated without further downstream activities; those are often required when e.g. spray drying provides fine ASD powders, which must be further densified via dry compaction before compression to tablets or multiparticulate mini-tablets.7
Re-crystallization of the API in an ASD would eliminate the improved dissolution behavior. Moisture reduces the glass transition temperature Tg of the ASD system and causes a plasticizing effect, which increases the molecular mobility of ASD and the risk of crystallization.6 To reduce the risk of recrystallization of the API, moisture must be excluded as much as possible.
2. Multiparticulate Solid Dispersions Including Solubilizer
Solid dispersions consisting of a poorly water-soluble or insoluble drug and an O/W surfactant are also used to improve drug solubility, adsorption, and bioavailability. The active ingredient is combined with the surfactant to form a solubilizer-drug matrix (Figure 4).
Coming into contact with water or gastrointestinal fluids, micelles are formed, taking up a water-insoluble drug into their lipophilic core. This situation is well known to everybody familiar with dishwashing and using a washing agent.
E.g. poloxamer type surfactants are known as efficient solubilizers. Poloxamers are block copolymers of poly(ethylene oxide) and poly(propylene oxide), which have an amphiphilic character and useful association and adsorption properties emanating from this (Figure 5).10
Solubilizer-based solid dispersions can be provided in the form of micropellets made with fluidized bed technologies such as fluid bed spray agglomeration, hotmelt pelletization (Glatt MicroPx process, Glatt ProCell process) or prilling technology.
3. Products with Multiparticulate Amorphous Solid Dispersions and Solid Dispersions including Solubilizer
Multiparticulates such as pellets and micropellets represent promising technology platforms, providing advantages when compared to monolithic oral dosage forms like tablets and capsules (Figure 6). These approaches can achieve better compliance for children and the elderly. Various drug products can be made from one multiparticulate form – capsules, oral liquids, sachets, stickpacks, sipping straws, fixed-dose combinations, and multiple unit pellet system (MUPS) tablets – offering interesting perspectives for life cycle management of an active compound.
Conclusion
New concepts for patient-centric solutions are possible. Small particles are easily administered via nasal and gastrointestinal tubes. Moreover, the pellet concept facilitates the processing of highly potent compounds to an extraordinary degree. Increasing solubility through these strategies can overcome a host of challenges:
- Poor water-soluble drugs can be transferred into amorphous solid dispersions (ASD) or solid dispersions (SD) including a solubilizer going along with a considerable improvement of solubility and bioavailability.
- Individually tailored drug release profiles for any API and site-specific drug delivery allow for an optimized and safe therapeutic effect.
- Viable process technology platforms are in place to realize even challenging drug product ideas.
- Even high-dosed drugs are presented in a palatable form for children and the elderly.
Case Studies: Examples of Different Solubility Strategies in Action
Case Study 1: Boosting the solubility of Nifedipine with ASD technology
Nifedipine, a dihydropyridine compound, exhibits a very low water solubility of 5 mg/L whereas the dose ranges up to 90 mg in controlled release preparations.
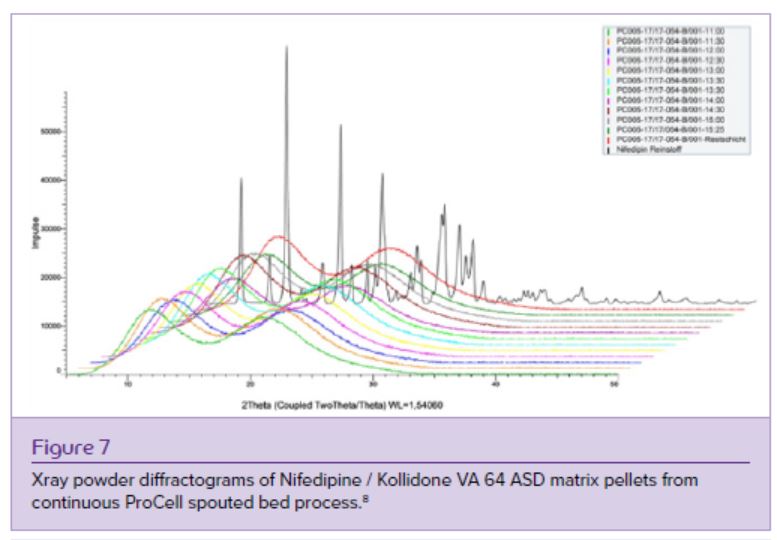
ASD layered and matrix pellets containing Nifedipine and Kollidon VA 64 (vinylpyrrolidone- vinyl-acetate-copolymer) in a ratio of 1/1.5 were generated using different fluidized bed technologies. The compounds were dissolved in acetone with a solids content of 30% w/w. Microcrystalline cellulose pellets Cellets 500 were used as inert starter beads for the drug layering process, for which a 6” Wurster fluidized bed unit with an air distribution plate type C was used (Figure 3A). 1 kg of Cellets 500 were layered with 4 kg of ASD liquid resulting in ASD layered pellets with a theoretical drug load of 21,8% w/w. ASD matrix pellets with a theoretical drug load of 40% w/w were produced with a Glatt ProCell 5 continuous spouted bed process (Figure 3B) equipped with a zig-zag air sifter for a continuous discharge of well-sized pellets. Samples for xray control of the amorphous state were taken every 30 min from the continuous process over 4,5 hrs to demonstrate the amorphous character of the product (Figure 7). Xray powder diffraction results proved stability of the products over two years stored at room temperature.
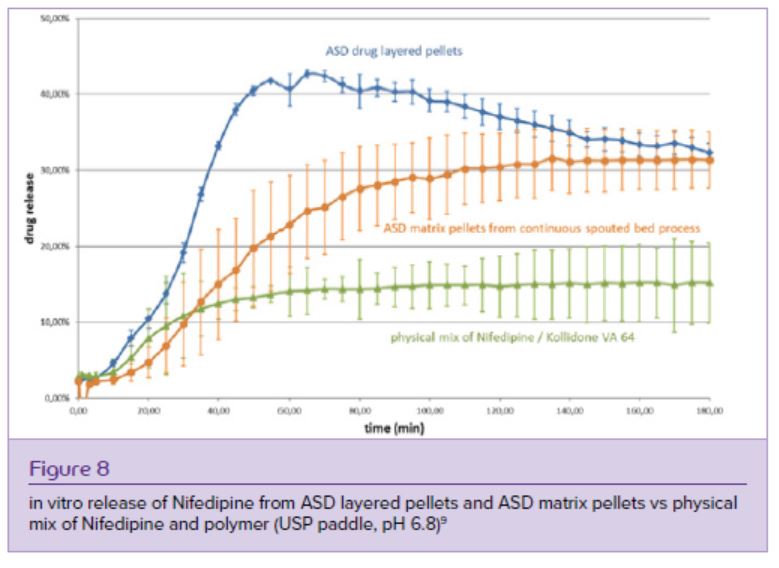
Nifedipine ASD pellets obtained with both technologies provided an approximately two-fold higher final drug release than the physical blend of Nifedipine and Kollidone VA 64. The ASD drug layered pellets dissolved faster than the ASD matrix pellets and showed a more pronounced supersaturation (Figure 8) – this finding is probably due to the lower drug load of the drug-layered pellets (21,8% w/w) compared with the matrix pellets (40% w/w).
Fluidized bed ASD layering and spouted bed ASD matrix pelletization technology proved to be promising platform technologies for manufacturing of stable Nifedipine/Kollidon VA 64 ASD products with good drug release performances. Resulting spherical ASD core pellets (Figure 9) are an ideal substrate for coating applications, which can provide taste-masking for bitter drugs or gastroresistant coating for acid-labile drugs, which would be degraded in the acidic medium of the stomach.
Case Study 2: Taste-masked Clarithromycine SD micropellets – making the insoluble drug soluble and hiding its bitter taste
Antibiotic drug Clarithromycine exhibits an extremely low water solubility of 0,33 mg/L.11 A 500 mg Clarithromycine dose therefore is 1667 times larger than the drug quantity soluble in water. In order to achieve a better solubility in water and to guarantee adequate bioavailability, a solid dispersion (SD) including poloxamer 188 as a solubilizer and polyvinylpyrrolidone (PVP K25) as a binder is prepared.
For a pediatric oral Clarithromycine liquid for children, the extremely bitter-tasting drug is transferred into SD micropellets using the MicroPx fluidized bed pelletization technology.
To manufacture Clarithromycine SD micropellets 200 – 400 μm, an aqueous dispersion containing micronized Clarithromycine, poloxamer 188 and PVP is prepared and sprayed into an initially empty MicroPx™ fluidized bed equipped with bottomspray nozzles; starter beads must not be used for this technology. When the particles reach the desired particle size, they are continuously removed from the MicroPx fluid bed process by way of a zigzag-type air sifter (Figure 11). Offering the advantage of continuous operation, the micropellets are produced with a high product yield > 95%. The production capacity of a midsized commercial MicroPx apparatus is 10 kg/h = 240 kg/day = 72 tons/year in the 7/24 shift mode within 300 working days.

The matrix micropellets contain 70% Clarithromycine, 18% poloxamer 188 and 12% polyvinylpyrrolidone K30. With the solubilisation approach, the huge difference between the extremely low water solubility of 0,3 mg/L and the dose range from 250 – 500 mg is overcome successfully going along with excellent bioavailability.
In a second processing step, the Clarithromycine SD micropellets are coated with a water-soluble HPMC-based seal coat in order to avoid drug migration into the final tastemasking film consisting of Eudragit L 30 D-55, triethylcitrate, glycerolmonostearate and Tween 80. The coatings are applied with the Wurster (bottomspray) fluidized bed technology. The coated micropellets are smaller than 500 μm to ensure a pleasant mouth feel; the drug is released immediately at pH 6.8 (Figure 10). Over a period of 14 days, efficient taste masking in a water-based oral suspension is achieved.12,13,15
Case Study 3: Improving the water solubility of CBD with solubilizer-based solid dispersion
Cannabidiol (CBD) is a lipophilic compound with a poor water solubility of 13 mg/L; the oral bioavailability of CBD is low and unpredictable.14 Formulation approaches aim for improving the solubility and bioavailability of the compound applied as an oral solid dosage form.
Solid dispersions of CBD and poloxamer 188 were prepared from a melt of the compounds, which was transferred into SD matrix pellets using fluid bed Wurster technology. For comparison reasons, CBD powder was hot-melt granulated with a poloxamer 188 melt using the same technology (Figure 12).
The in vitro CBD release from the hot melt granulation process was found to be faster, but less complete than the CBD release from the matrix pellets made from a hot melt of both compounds (Figure 11). In the CBD/poloxamer 188 matrix, the contact of the active compound and the surfactant is considered to be more advantageous than in the hot melt granule, as both CBD and poloxamer 188 were molten and transferred into a homogenous matrix pellet from there.15
References
1. Kawabata, Y.; Wada, K.; Nakatani, M.; Yamada, S.; Onoue, S. Formulation design for poorly water-soluble drugs based on biopharmaceutical classification systems: basic approach and practical applications; Int. J. Pharm. 2011, 420 (1), 1 – 10
2. T. Vasconcelos, B. Sarmento, P. Costa; Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs; Drug Discovery Today 2007, 12, no. 23, 1068 – 1075 3. C. Vo, C. Park, B. Lee. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs, European Journal of Pharmaceutics and Biopharmaceutics 2013, 85, no. 3, 799 – 813
4. J. Breitenbach. Melt extrusion: from process to drug delivery technology, European Journal of Pharmaceutics and Biopharmaceutics 2002, vol. 54, no. 2, 107 – 117
5. US Patent 4,892,730; A. Hegasy, Bayer AG, 1990
6. Bhujbal, S.V.; Mitra, B.; Jain, U.; Gong, Y.; Agrawal, A.; Karki, S.; Taylor, L.; Kumar, S.; Zhou, Q. Pharmaceutical solid dispersion: A review of manufacturing strategies, Acta Pharmaceutica Sinica, 2021
7. L. Weuts et al; Physicochemical properties of the amorphous drug etc, Journal of Pharmaceutical Sciences 2011, vol. 100, no. 1, 260 – 274
8. Neuwirth, M. Versuchsbericht: Entwicklung einer Methode zur kontinuierlichen Produktion einer Amorphen Solid Dispersion (ASD) in der Wirbelschicht. Glatt Pharmaceutical Services/Univ. Bonn, 2017
9. Neuwirth, M.; Grave, A.; Pöllinger, N.; Wagner, Karl G. Fluidized and spouted bed for the preparation of directly processable amorphous solid dispersions; 12th World meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology, Vienna, 2020
10. Bodratti, A.; Alexandridis P. J. Funct. Biomater. 2018 9 (1),
11 ; reprinted with permission from Bedrov, D. et al, J. Chem. Theory Comput. 2006, 2, 598 – 606 11. Drug Bank, accession number DB 01211; https://go.drugbank.com
12. Pöllinger, N.; Drug product development for older adults – multiparticulate formulations in Developing Drug Products in an Aging Society, ed. S. Stegemann, AAPS/Springer 2016
13. Prasch, A.; Luy, B.; Pöllinger, N.; Struschka, M.; Schwarz, F.X. EP 1 631 373 B1, 2008 14. Zhornitsky, S.; Potvin, S. Cannabidiol in humans – the quest for therapeutic targets, Pharmaceuticals 2012, 5, 529 – 555 15. Internal Glatt documents of various years, which have not been released to the public