 Jennifer Putnam Perrigo
Jennifer Putnam Perrigo
Katherine Ulman KLU Consulting
David Schoneker Black Diamond Regulatory Consultants
Kim Beals IPEC-Americas
The COVID-19 pandemic caused tremendous supply chain disruptions across practically every industry segment. The pharmaceutical industry was impacted by global trade restrictions as quarantine protocols limited manufacturing and nations around the world limited exports of finished drug products and the materials needed to manufacture them. As these limitations began to lift, the global transportation infrastructure could not keep up with the rapid increase in trade, extending the supply chain crisis.
Qualifying multiple, interchangeable sources of excipients used in the manufacture of a drug product could reduce the potential impact of future global supply chain disruptions. However, careful consideration must be taken to ensure that unintended consequences do not result from limited evaluation of alternative sources.
There are other good business reasons that qualification of multiple excipient sources should be considered during the drug product lifecycle—from formulation development, through commercial manufacturing. Demand for the excipient may exceed the volume a single source can provide. A company manufacturing material for use as pharmaceutical excipients as well as food or cosmetic ingredients could decide to discontinue manufacture of their pharmaceutical grade and occasionally, lower-cost alternatives may relieve external pricing challenges.
As with any business decision, when the risks are not fully understood, the total cost of qualifying and maintaining multiple suppliers can be greater than perceived cost savings. While enticing, the potential cost savings may not be sustained if the lower cost alternative is less reliable or if the price of the material is increased after implementation. In fact, managing multiple suppliers could even increase total operational and quality costs. It is usually more effective for the drug product manufacturer to work with existing suppliers on business continuity and future needs— with the goal of building strong relationships and creating long term value for both parties.
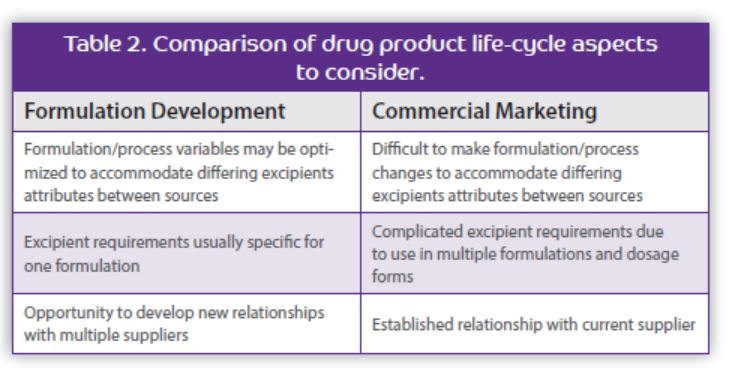
Qualification Considerations Successfully implementing an alternate supplier for an excipient, when needed, depends directly on a comprehensive risk-based qualification confirming the interchangeability of the alternate source excipient in the drug product formulation, which is often more technically challenging than expected. While regulatory requirements for approval of an alternate source excipient is typically much less than for an API, the selection and qualification can be more complex (refer to Table 1). The term “inactive” ingredient is somewhat misleading because excipients are necessary drug product components, often with attributes important for the manufacturing or quality of the drug product.

The excipient composition profile often includes other concomitant components that may be important for the excipient’s performance and could vary significantly between suppliers due to differences in raw materials and manufacturing processes. No two sources will ever be identical. It is important that the composition of the current source excipient is well understood, but this can be challenging for excipients used in marketed drug products if not considered during formulation development. Communication with the current supplier (not just the alternate supplier) is often necessary. Care should be taken to avoid damaging the relationship.
It is also important that the excipient’s critical material attributes are well understood. Ideally, Quality by Design (QbD) risk assessments and studies are performed to determine Critical Material Attributes (CMAs) and design space. When excipient attributes are not well understood (which is common for drug products commercialized 20+ years ago) or are expected to be variable, extensive evaluation of the functional performance of the alternate excipient source in the drug product formulation is necessary to ensure successful implementation (refer to Table 2). More extensive evaluations are needed when the excipient attributes are important for drug product manufacture or performance, especially if the attribute is not directly measurable in the excipient.
While alternate source excipients are not explicitly addressed in the FDA guidances for post-approval changes, commonly referred to as the “SUPAC (Scale-up and Post Approval Changes) guidances”, the FDA does provide a framework for classifying and determining the potential impact of implementing the alternate source. While there is usually minimal impact to drug product applications for alternate source excipients, unless there is a change to the specification or technical grade of the excipient, the SUPAC guidances require the assessment of any changes that could impact drug product performance. A change in the composition of the excipient may lead to a change in the drug product formulation, which could impact labeling.
In a 2022 Excipient World presentation,1 Sam Raney indicated that Q1 (components in a product), Q2 (composition of a product) and Q3 (arrangement of matter in a product) differences should each be assessed for their impact on bioequivalence of topical product formulations. The Q1/2/3 could be impacted by an alternate source excipient because the excipient composition and other attributes impacting the excipient function could vary between suppliers.
Equivalency and Interchangeability
For most excipients, it is practically impossible for any two sources to be identical. The concepts of equivalency and interchangeability are important to understand within the context of alternate source qualification, because the alternate source excipient must do the exact same thing in the drug product as the current source—but doesn’t necessarily need to be identical in all aspects to perform acceptably. Equivalence is demonstrated when the excipient meets predefined material attribute requirements; interchangeability is demonstrated when the excipient has been confirmed to perform as required in the drug product.
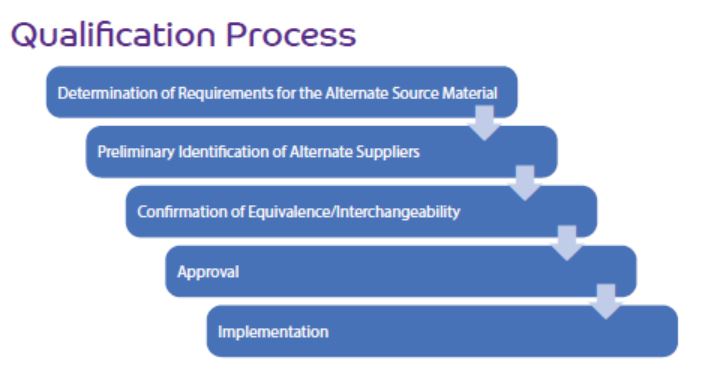
The qualification process consists of a stepwise risk assessment performed by a cross-functional team with expertise in raw materials, formulation, quality, commercial operations, procurement, regulatory affairs, stability, and other technical subject matters based on the drug product application. The requirements for the alternate source excipient are defined by the team, and potential alternate sources are compared for preliminary selection. The team develops and executes a testing strategy for experimental confirmation of equivalence and interchangeability in the drug product formulation. Approval and implementation of the alternate source excipient entail formal change control documentation and activities such as process validation and on-going monitoring.
Defining Alternate Source Requirements
Defining the alternate source excipient requirements is an important step that needs to be performed by the cross-functional team. The excipient specification alone should not be assumed to adequately capture all of the requirements. Conversely, inclusion of a test for an attribute on a specification does not necessarily indicate the criticality of the attribute. Comparison of specification criteria alone is usually not sufficient to predict equivalency or interchangeability of excipients that are important for the drug product manufacture or performance. Sharing the specification directly with potential suppliers without context for usage may result in significant delays due to the material not demonstrating equivalent performance. The alternate source excipient requirements may be general attributes as well as specific and defined attributes. General attributes include quality attributes such as identification (e.g., corn starch vs. pregelatinized starch) and compliance to the compendial monograph. Some alternate source excipients may only need to meet general attribute type requirements. However, based on its function, many will have specific requirements, such as moisture content, particle size distribution, viscosity, or animal vs. plant-based, which may not be included in the excipient specification. In addition to the main function, an excipient may have unknown or undeclared functions.
As depicted in the figure to the right, some excipients may also have important material attributes that are not apparent or well characterized. Therefore, a risk assessment by the cross-functional team is essential to establishing these attributes. The potential for risk due to other important material attributes differs based on the nature and function of the excipient as well as the drug product application. If the alternate source excipient will be used in multiple applications, they should be considered separately, as the function of the excipient may not be the same for all applications.
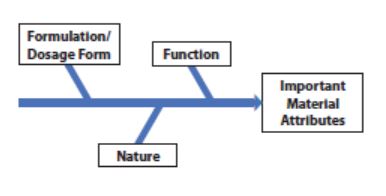
The nature of the excipient, such as natural vs. synthetic or single chemical entity vs. complex composition, can contribute to the potential for differences in attributes such as composition and batch-to-batch variation between sources. It is important that the full composition, including the presence of additives and residual processing aids, and the potential for batch-to-batch variation, is understood. In addition, the formulation robustness should be considered, including the history of deviations resulting in any raw material controls by the excipient manufacturer. The risk assessment should be documented. The cross functional team should develop a tool to both guide and document the risk assessment. Quality by Design risk assessment- type tools are helpful for ensuring thorough and consistent risk assessments; the IPEC QbD guide2 is a good reference for developing such tools.
Preliminary Selection
The alternate source selection process begins by considering business drivers such as market position, pricing, lead time and other legal and contractual obligations. The strength of the relationship between the excipient supplier and drug product manufacturing is important to consider. The supplier’s transparency, accessibility and engagement—as well as technical expertise—are vital to successful implementation, and should be strongly weighed.
The cross-functional team evaluates the alternate supplier’s technical and quality documentation as compared to the predefined requirements, with the goal of identifying one or more sources that are expected to be interchangeable. The rationale for the preliminary supplier selection should be documented. Potential risks to successful implementation should be identified and mitigation plans for all high risks, and medium risks (when possible) should be developed.
Confirmation of Interchangeability
The cross-functional team should develop a testing strategy based on the predefined requirements to confirm interchangeability of the alternate source excipient. The testing strategy should include testing alternate source material for the specification requirements and other important measurable attributes that may have been determined via the risk assessment.
Typically, at least three representative batches are evaluated, and the results compared to the historical data for the current material as well as the specification acceptance criteria. It is important to engage with the supplier regarding the potential for variation. If there is potential for seasonal or other known long-run variability, batches representative of the expected variation should be evaluated, if available. It is usually not practical or possible to test the potential extremes.
No other data beyond the raw material testing may be required, depending on the risk assessment and the excipient function and application, but evaluation under the actual conditions of use is usually necessary to confirm interchangeability.
While full scale GMP batches are usually necessary, informal or pilot scale batches or even single unit processes, such as granulation, may be sufficient to provide the information needed to confirm interchangeability. Usually only a single drug product batch is necessary as multi-batch trials are only likely to detect frequent or wide material variation. The drug product testing strategy should also be based on risk assessment. Even if not required for regulatory approval, pre-market stability should be considered, unless there is documented scientific justification that concurrent stability is sufficient. If the excipient is important for manufacturing, attributes such as content uniformity should be considered.
Approval and Implementation
While prior FDA approval is generally not required—except for change in grade or specification—formal internal change control is always required for an alternate source excipient and should include documentation of the alternate supplier selection, including the risk assessment, supplier selection rationale and confirmation of equivalence and interchangeability. Implementation of the alternate source excipient includes activities such as process validation and concurrent stability. The performance of the excipient should be monitored because trends due to attribute variation may not be revealed until the tenth (or 100th!) drug product batch utilizing the alternate source has been manufactured. When adverse performance trends are identified post-implementation, communication with the excipient supplier is critical for determining the cause.
Summary
Using alternate sources of excipients after drug product formulation and approval is often required to maintain adequate supply to support commercial drug product manufacturing. Selection, qualification, and implementation of alternate source excipients requires formal/documented change control. Excipients meeting the same compendial requirements may not be interchangeable in all drug product formulations. Risk assessment (aka QbD) approach is valuable for determination of material requirements. Equivalency and interchangeability must be confirmed in the context of the material usage. Finally, while regulatory requirements are minimal (assuming no change to the excipient grade or specification), successful implementation is often technically challenging.
Equivalence Example
A screwdriver can be defined as a tool with a handle, shank and tip designed to drive screws.
None of the screwdrivers shown in this image are identical because there are differences between the handles, shanks, and tips. Whether any are equivalent depends on the attribute requirements.
One requirement might be for it to have a slot-head to fit the screw. All of the screwdrivers that fit slot-head screws are equivalent in regard to this attribute. If a second requirement is determined to be long shank length due to the position of the screw, only screwdrivers with slot-tips and long shanks meet both requirements. The slot-tip screwdrivers with very short shanks would no longer be considered equivalent.
CMA Example
Xanthan gum functions as a suspension agent in oral suspension products and as an emollient in oral lozenges. A critical material attribute for the suspension application should consider viscosity, but viscosity would not be a consideration when used as an emollient.
References
1. 2022 Excipient World presentation entitled The Critical Impact of Excipients on the Physicochemical and Structural Characteristics of Topical Drug Products, Sam Raney, FDA Associate Director for Science, Chief Scientific Advisor for Topical Drug Products.
2. The International Pharmaceutical Excipient Council Incorporation of Pharmaceutical Excipients into Product Development using Quality-by-Design (QbD) Guide, 2020.